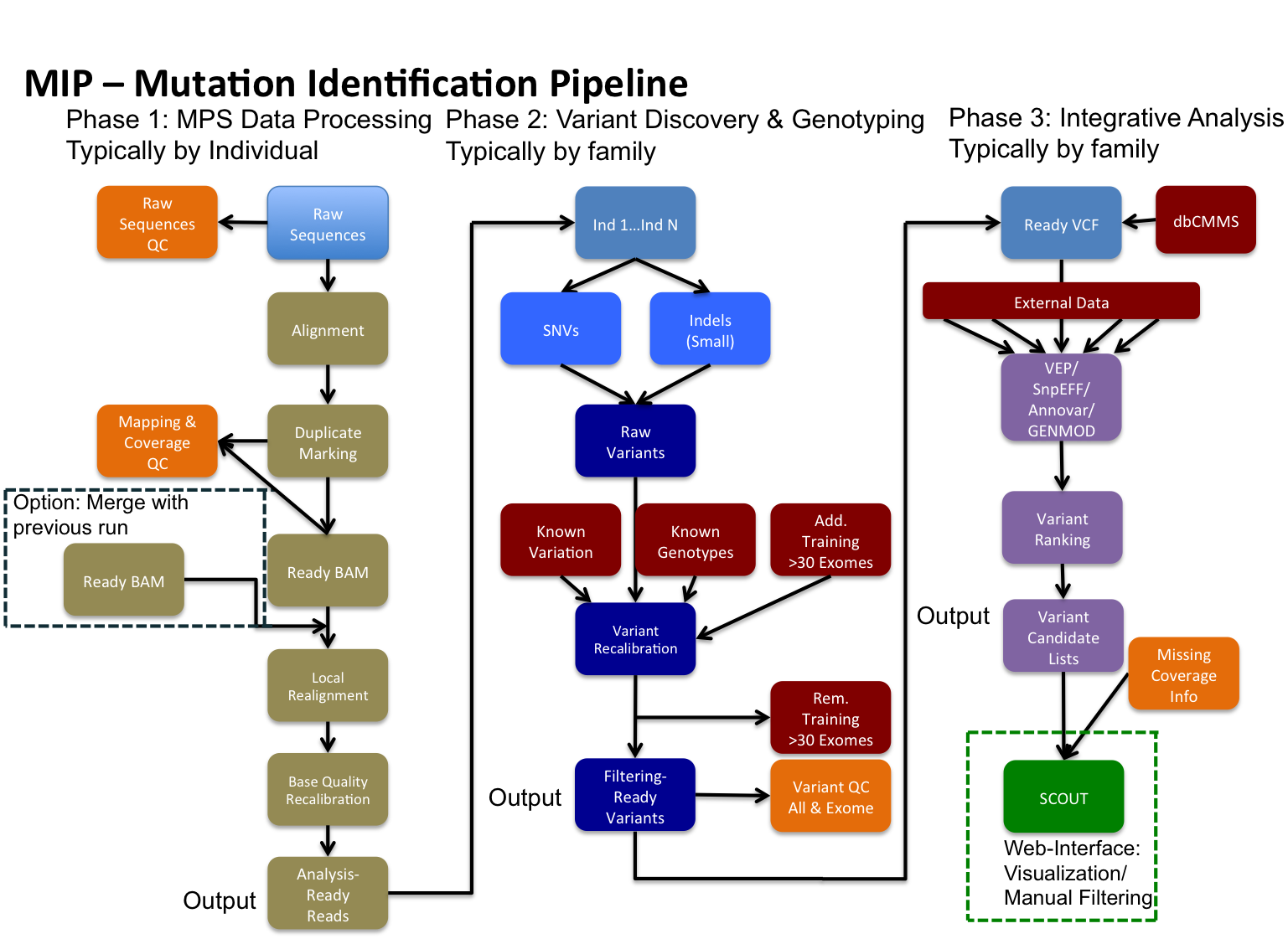
MIP - Mutation Identification Pipeline¶
MIP enables identification of potential disease causing variants from sequence data.
Overview¶
MIP performs whole genome or target region analysis of sequenced single-end and/or paired-end reads from the Illumina platform in fastq(.gz) format to generate annotated ranked potential disease causing variants. MIP performs QC, alignment, coverage analysis, variant discovery and annotation, sample checks as well as ranking the found variants according to disease potential with a minimum of manual intervention. MIP is compatible with Scout and Puzzle for visualization of identified variants. MIP has been in use in the clinical production at the Clinical Genomics facility at Science for Life Laboratory since 2014.
Features¶
- Installation
- Simple automated install of all programs using conda/SHELL via supplied install script
- Autonomous
- Checks that all dependencies are fulfilled before launching
- Builds/Prepares/downloads references and/or files missing before launching
- Decompose and normalise reference(s) and variant vcf(s)
- Splits and merges files/contigs for samples and families when relevant
- Automatic
- A minimal amount of hands-on time
- Tracks and executes all module without manual intervention
- Creates internal queues at nodes to optimize processing
- Minimal IO between nodes and login node
- Flexible:
- Design your own workflow by turning on/off relevant modules
- Restart an analysis from anywhere in your workflow
- Process one, or multiple samples using the module(s) of your choice
- Supply parameters on the command line, in a pedigree file or via config files
- Simulate your analysis before performing it
- Redirect each modules analysis process to a temporary directory (@nodes or @login)
- Limit a run to a specific set of genomic intervals
- Use multiple variant callers and annotation programs
- Optionally split data into clinical variants and research variants
- Fast
- Analyses an exome trio in approximately 4 h
- Analyses a X-ten sequenced genome in approximately 21 h
- Rapid mode analyzes a WGS sample in approximately 4 h using a data reduction and parallelization scheme
- Traceability
- Track the status of each modules through dynamically updated status logs
- Recreate your analysis from the MIP log or generated config files
- Logs sample meta-data and sequence meta-data
- Logs version numbers of softwares and databases
- Checks sample integrity (sex and relationship)
- Test data output existens and integrity using automated tests
- Annotation
- Gene annotation
- Summarise over all transcript and output on gene level
- Transcript level annotation
- Separate pathogenic transcripts for correct downstream annotation
- Annotate all alleles for a position
- Split multi-allelic records into single records to ease annotation
- Left align and trim variants to normalise them prior to annotation
Annotate coverage across genetic regions
Extracts QC-metrics and stores them in YAML format
- Standardized
- Use standard formats whenever possible
- Visualization
- Ranks variants according to pathogenic potential
- Output is directly compatibel with Scout and Puzzle
Example Usage¶
perl mip.pl -pMosaikBuild 0 -configFile 1_config.yaml
Getting Started¶
Installation¶
MIP is written in Perl and therfore requires that Perl is installed on your OS (See Installation).
Change log (See Change Log)
Prerequisites¶
MIP will only require prerequisites when processing a modules that has dependencies (See Setup). However, some frequently used sequence manipulation tools e.g. samtools, PicardTools, Bedtools are probably good to have in your path.
Meta-Data¶
Meta data regarding the pedigree, gender and phenotype should be supplied for the analysis.
- Pedigree file (PLINK-format; See Pedigree File & MIP´s github repository).
- Configuration file (YAML-format; See Dynamic Configuration File & MIP´s github repository).
Usage¶
MIP is called from the command line and takes input from the command line (precedence), a config file (yaml-format) or falls back on defaults where applicable.
Lists are supplied as comma separated input, repeated flag entries on the command line or in the config using the yaml format for arrays.
Note
List or repeated entries need to be submitted with the same order for each element across all supplied lists.
Only flags that will actually be used needs to be specified and MIP will check that all required parameters and dependencies (for these flags only) are set before submitting to SLURM.
Program parameters always begins with “p” followed by a capital letter. Program parameters can be set to “0” (=off), “1” (=on) and “2” (=dry run mode). Any program can be set to dry run mode and MIP will create sbatch scripts, but not submit them to SLURM for these modules. MIP can be restarted from any module, but you need to supply previous dependent programs in dry run mode to ensure proper file handling.
MIP will overwrite data files when reanalyzing, but keeps all “versioned” sbatch scripts for traceability.
MIP allows individual target file calculations if supplied with a pedigree file or config file containing the supported capture kits for each sample.
You can always supply perl mip.pl -h to list all available parameters and
defaults.
Example usage:
$ perl mip.pl -f 3 -sampleid 3-1-1A,3-2-1U -sampleid 3-2-2U -pFQC 0 -pMosaikBuild 2 -pMosaikAlign 2 -c 3_config.yaml
This will analyze family 3 using three individuals from that family and begin the analysis with programs after MosaikAlign and use all parameter values as specified in the config file, except those supplied on the command line, which has precedence.
Input
It is recommended to use MIP’s naming convention for input Fastq files to accurately and automatically handel individual runs and lanes (See Setup).
Fastq files (gziped/uncompressed) should be place within the -inFilesDirs as -inFilesDirs {PathToInFileDir}=sampleID.
Note
MIP will automatically compress any non gzipped files if -pGZip is enabled.
All files ending with .fastq or .fast.gz will be included in the run.
MIP scripts locations are specified by -inScriptDir.
All references and template files should be placed directly in the reference
directory specified by -referencesDir, except for ANNOVAR db files, which
should be located in annovar/humandb.
Output
Analyses done per individual is found under respective sampleID subdirectory and analyses done including all samples can be found under the family directory.
Sbatch Scripts
MIP will create sbatch scripts (.sh) and submit them in proper order with
attached dependencies to SLURM. These sbatch script are placed in the output
script directory specified by -outScriptDir. The sbatch scripts are versioned
and will not be overwritten if you begin a new analysis. Versioned “xargs” scripts will also
be created where possible to maximize the use of the cores processing power.
Data
MIP will place any generated datafiles in the output data directory specified by
-outDataDir. All datatfiles are regenerated for each analysis. STDOUT and
STDERR for each program is written in the <program>/info directory prior to
alignment and in the <aligner>/<program>info directory post alignment.
Analysis Types
Currently, MIP handles WES -at sampleID=wes, WGS -at sampleID=wgs or Rapid analysis -at sampleID=rapid for acute patient(s).
The rapid analysis requires BWA_MEM and selects the data that overlaps with the regions supplied with
the -bwamemrdb flag. MIP will automatically detect if the sequencing run is single-end or paired-end/interleaved paired-end
and the length of the sequences and automatically adjust accordingly.
Note
In rapid mode; Sort and index is done for each batch of reads in the BWA_Mem call, since the link to infile is broken by the read batch processing.
However pPicardToolsSortSam should be enabled to ensure correct fileending and merge the flow to ordinary modules.
Project ID
The -projectID flag sets the account to which core hours will be allocated in SLURM.
Aligner
Currently MIP officially supports two aligners Mosaik and BWA, but technically supports any aligner that outputs BAM files. Follow the instructions in Adding a new program to add your own favorite aligner.
Log
MIP will write the active analysis parameters and STDOUT to a log file located in:
{OUTDIRECTORY}{FAMILYID}/{MIP_LOG}/{SCRIPTNAME_TIMESTAMP}
Information, such as infile, programs, outdatafiles etc, for each analysis run is dynamically
recorded in the a yaml file determined by the -sampleInfoFile flag. Information in the sampleInfo
file will be updated in each analysis run if identical records are present and novel entries are added.
The sampleInfo file is used in QCCollect to extract relevant qc metrics from the MPS analysis.
Pipeline WorkFlow
This is an example of a workflow that MIP can perform (used @CMMS).